Some of our favorite papers from 2021
 Image credit: Clipartmax
Image credit: ClipartmaxPreface
First, let’s go over the three caveats of this paper review post.
Caveat #1: Although the list of the papers and the reasons for choosing them are an amalgam of the opinions expressed by the lab members, the post itself is narrated in a single voice, and as such is much more subjective.
Caveat #2: Science is not a competition, and thus the concept of ranking or selecting a top-N list is not exactly well suited for discussion of scientific publications. Instead, this should be viewed as selection of works that stood out to us due to their relevance to what we work on, ingenuity of the methods, and overall style. In other words, this is a short snippet of what we really enjoyed reading, rather than some arbitrary ranking.
Caveat #3: As a corollary to Caveat #2 the list of the papers will not have an actual ranking between the works selected, and rather be presented based on the thematic grouping. This should make for an easier discussion and summarization of our opinions.
Caveat #4: We actively decided to avoid SARS-CoV-2 specific papers (we are saving a similar post just on SARS-CoV-2 for a later date).
Caveat #5: We will primarily focus on papers describing software and computational methods.
Now, with the layout described and disclaimers taken care of let’s dive into some of our favorite papers from 2021.
Table of contents
- Pangenomes
- Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. [DOI:10.1126/science.abg8871, bioRxiv:10.1101/2020.12.04.412486]
- ODGI: understanding pangenome graphs. [bioRxiv:10.1101/2021.11.10.467921]
- Read Until
- Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. [DOI:10.1038/s41587-020-0731-9, bioRxiv:10.1101/2020.02.03.931923]
- Readfish enables targeted nanopore sequencing of gigabase-sized genomes. [DOI:10.1038/s41587-020-00746-x, bioRxiv:10.1101/2021.12.01.470722]
- SquiggleNet: real-time, direct classification of nanopore signals. [DOI:10.1186/s13059-021-02511-y, bioRxiv:10.1101/2021.01.15.426907]
- Assembly
- Towards complete and error-free genome assemblies of all vertebrate species. [DOI:10.1038/s41586-021-03451-0, bioRxiv:10.1101/2020.05.22.110833]
- Robust haplotype-resolved assembly of diploid individuals without parental data. [arXiv:2109.04785]
- Minimizer-space de Bruijn graphs: Whole-genome assembly of long reads in minutes on a personal computer. [DOI:10.1016/j.cels.2021.08.009, bioRxiv:10.1101/2021.06.09.447586]
- Meta -omics & Microbiome
- Microbiome-based disease prediction with multimodal variational information bottlenecks. [bioRxiv:10.1101/2021.06.08.447505]
- Fast and accurate metagenotyping of the human gut microbiome with GT-Pro. [DOI:10.1038/s41587-021-01102-3, bioRxiv:10.1101/2020.06.12.149336]
- Gramtools enables multiscale variation analysis with genome graphs. [DOI:10.1186/s13059-021-02474-0]
- Nick’s pick
- The Labeled Direct Product Optimally Solves String Problems on Graphs. [arXiv:2109.05290]
Pangenomes [2 papers]
- Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. [DOI:10.1126/science.abg8871, bioRxiv:10.1101/2020.12.04.412486]
 |
|---|
| Fig. Abstract from Sirén, Jouni, Jean Monlong, Xian Chang, Adam M. Novak, Jordan M. Eizenga, Charles Markello, Jonas A. Sibbesen et al. “Pangenomics enables genotyping of known structural variants in 5202 diverse genomes.” Science 374, no. 6574 (2021): abg8871. |
- ODGI: understanding pangenome graphs. [bioRxiv:10.1101/2021.11.10.467921]
 |
|---|
| Fig. 1 from Guarracino, Andrea, Simon Heumos, Sven Nahnsen, Pjotr Prins, and Erik Garrison. “ODGI: understanding pangenome graphs.” bioRxiv (2021). |
In order to study the genomic diversity both within and across species we need to build references that account for variation. With this thesis in mind emergence and growth of pangenome based tools is clear.
The first paper introduces Giraffe — a short read mapper that uses pangenome reference for mapping the reads. Rationale behind building this tool is two-fold: on one hand, short read sequencing is still the cheapest form of sequencing tech, on the other hand, we have started accumulating large catalogs of genomic diversity via long read sequencing allowing us to construct pangenomes. At the core of this tool lies the work on graph Burrows-Wheeler transform and construction of haplotype aware pangenome indices. While most of the paper is focused on read mapping to human pangenome authors perform some simulated experiments with a yeast pangenome. Given the noticeable difference in the pangenome graphs it remains an open question to fully extend principles and ideas of pangenome reference mapping to microbial world.
The second paper introduces ODGI — a toolkit for working with pangenome graphs. Given the importance of pangenomes for the state of the art comparative genomics studies emergence of a toolkit for efficient manipulation of the underlying data types was expected. Effectively authors aim to provide BEDtools-like experience for pangenome graph manipulation. It is clear that with the growth of use of the pangenome representations our ability to manipulate them efficiently will become key to any further scientific advances, and thus the quality of our toolkits will determine the pace of comparative genomics in years to come.
Read Until [3 papers]
- Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. [DOI:10.1038/s41587-020-0731-9, bioRxiv:10.1101/2020.02.03.931923]
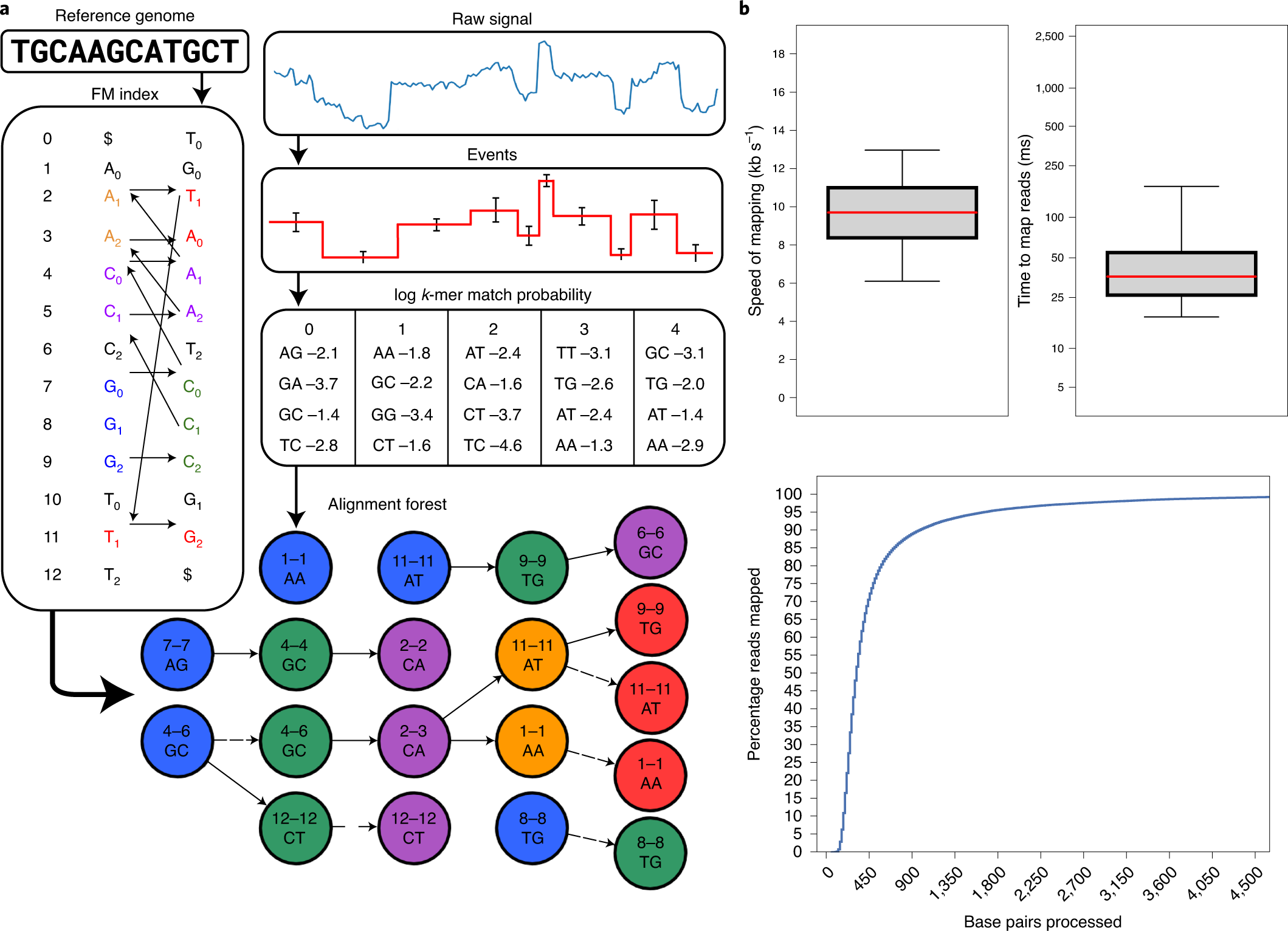 |
|---|
| Fig. 1 from Kovaka, Sam, Yunfan Fan, Bohan Ni, Winston Timp, and Michael C. Schatz. “Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED.” Nature Biotechnology 39, no. 4 (2021): 431-441. |
- Readfish enables targeted nanopore sequencing of gigabase-sized genomes. [DOI:10.1038/s41587-020-00746-x, bioRxiv:10.1101/2021.12.01.470722]
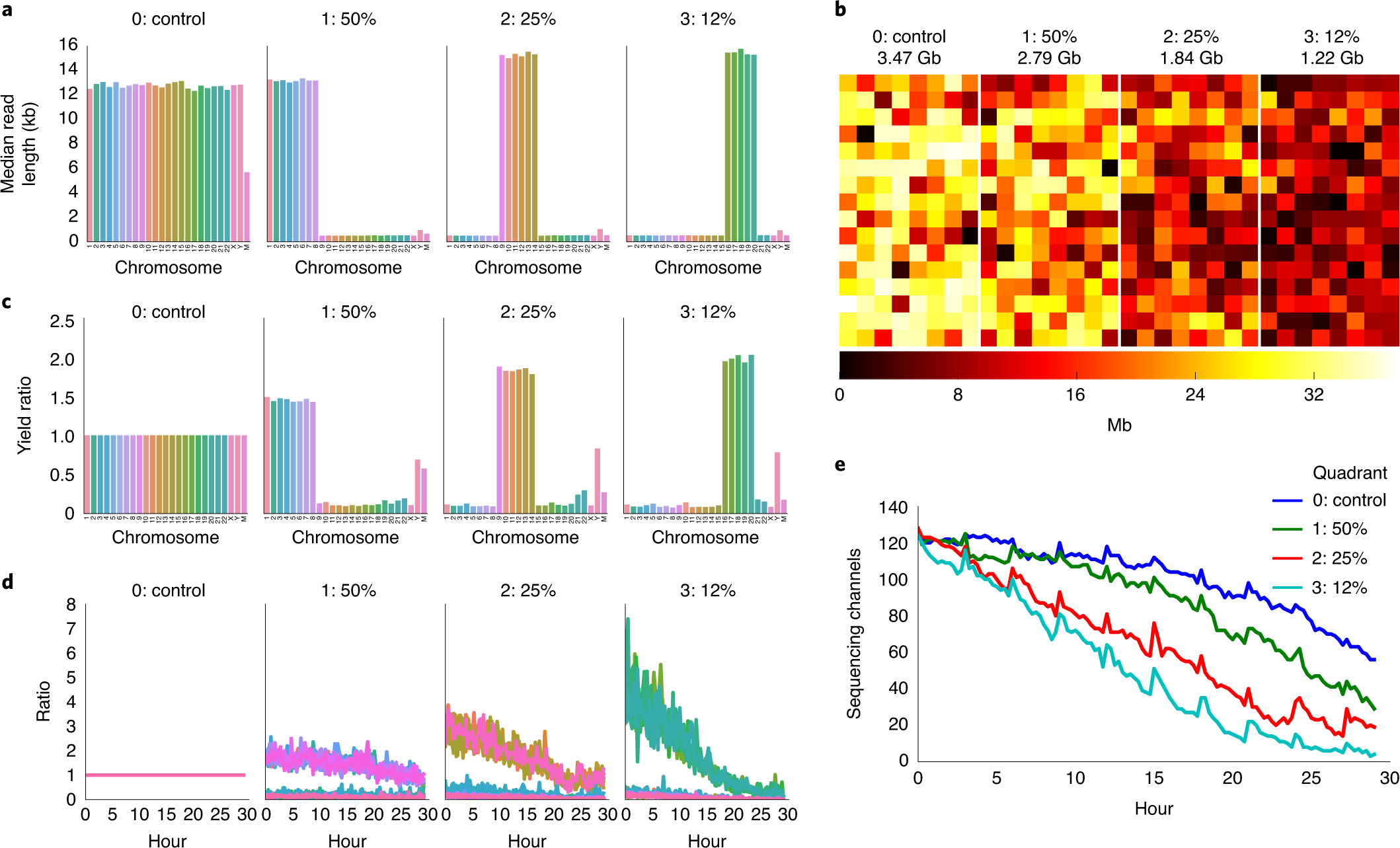 |
|---|
| Fig. 1 from Payne, Alexander, Nadine Holmes, Thomas Clarke, Rory Munro, Bisrat J. Debebe, and Matthew Loose. “Readfish enables targeted nanopore sequencing of gigabase-sized genomes.” Nature biotechnology 39, no. 4 (2021): 442-450. |
- SquiggleNet: real-time, direct classification of nanopore signals. [DOI:10.1186/s13059-021-02511-y, bioRxiv:10.1101/2021.01.15.426907]
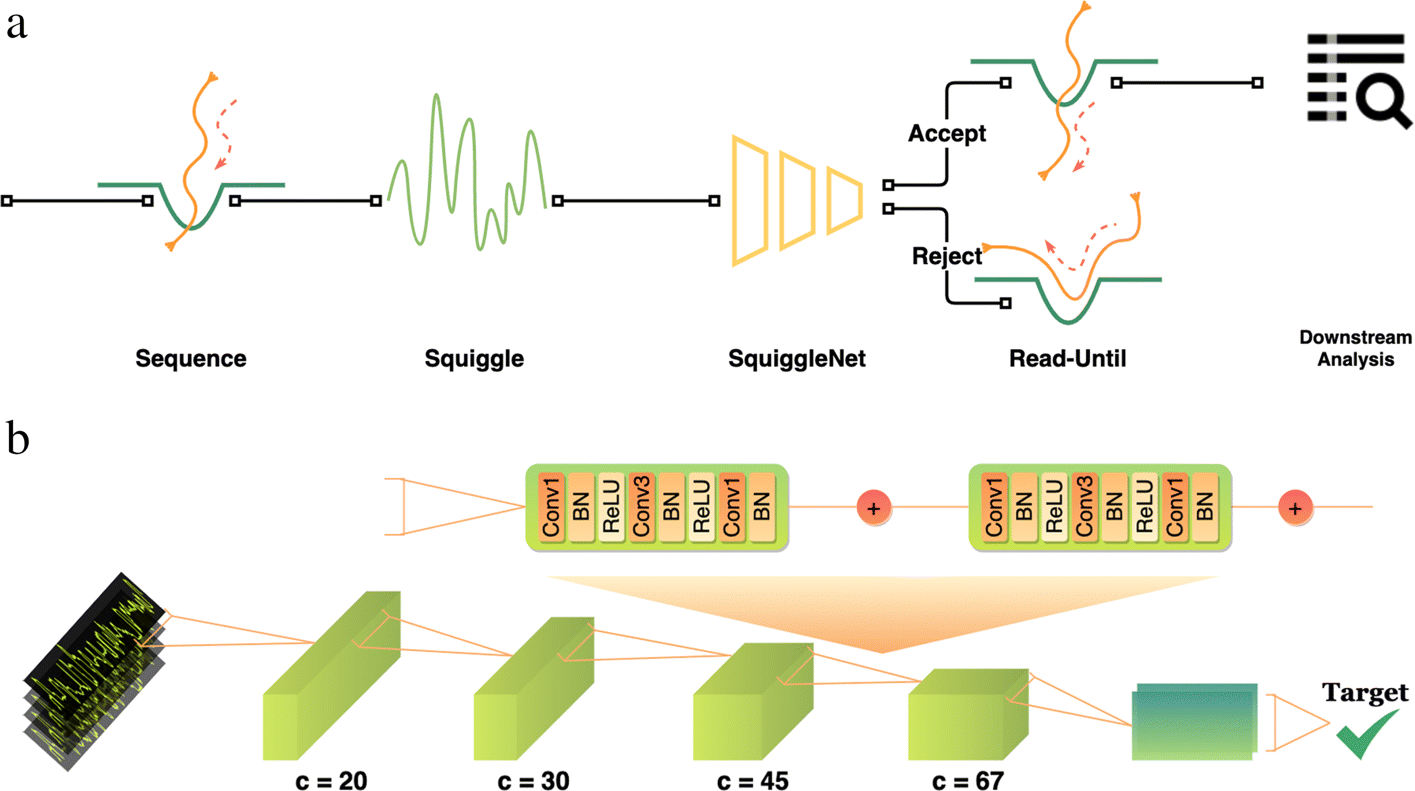 |
|---|
| Fig. 1 from Bao, Yuwei, Jack Wadden, John R. Erb-Downward, Piyush Ranjan, Weichen Zhou, Torrin L. McDonald, Ryan E. Mills et al. “SquiggleNet: real-time, direct classification of nanopore signals.” Genome biology 22, no. 1 (2021): 1-16. |
These three papers focus on taking advantage of real-time Read Until technology from Oxford Nanopore Technologies sequencing. Read Until method allows reads to be discarded while sequencing is still happening.
This approach to sequencing presents a unique set of advantages. First, Read Until based methods allow retaining the benefits of untargeted sequencing, since read ejection is driven by computational approaches and does not require a targeted library preparation step. Second, usage of Read Until can improve cost efficiency of sequencing, in silico targeting can improve coverage for the target organism(s) while still using the general purpose reagents and sequencer. Third, usage of Read Until can help with the removal of human genomic data from patient derived microbial samples, which in turn addresses both contamination and privacy issues of microbial sequencing reads in host derived samples.
In order to unlock those benefits, methods using Read Until API must be time-efficient. Thus, the development of new approaches to read classification is warranted. Each of the above papers takes a different approach to this problem. UNCALLED and SquiggleNet operate directly on the electrical current signals from the ONT device, while Readfish performs base-calling that is accelerated by the use of GPUs. Furthermore, while UNCALLED and Readfish rely on read mapping as their classification method (Readfish also can use Centrifuge instead of minimap2), SquiggleNet takes an end-to-end approach with a 1D-ResNet-based classifier.
Regardless of the approach all methods have their own computational footprint based limitations. However, it is clear that the development of improved classifiers for use with Read Until became and important topic in 2021. Thus, it’s exciting to see what novel discoveries will be brought to us in 2022 by usage of these tools, and what novel toolkits will get inspired by the Read Until.
Assembly [3 papers]
- Towards complete and error-free genome assemblies of all vertebrate species. [DOI:10.1038/s41586-021-03451-0, bioRxiv:10.1101/2020.05.22.110833]
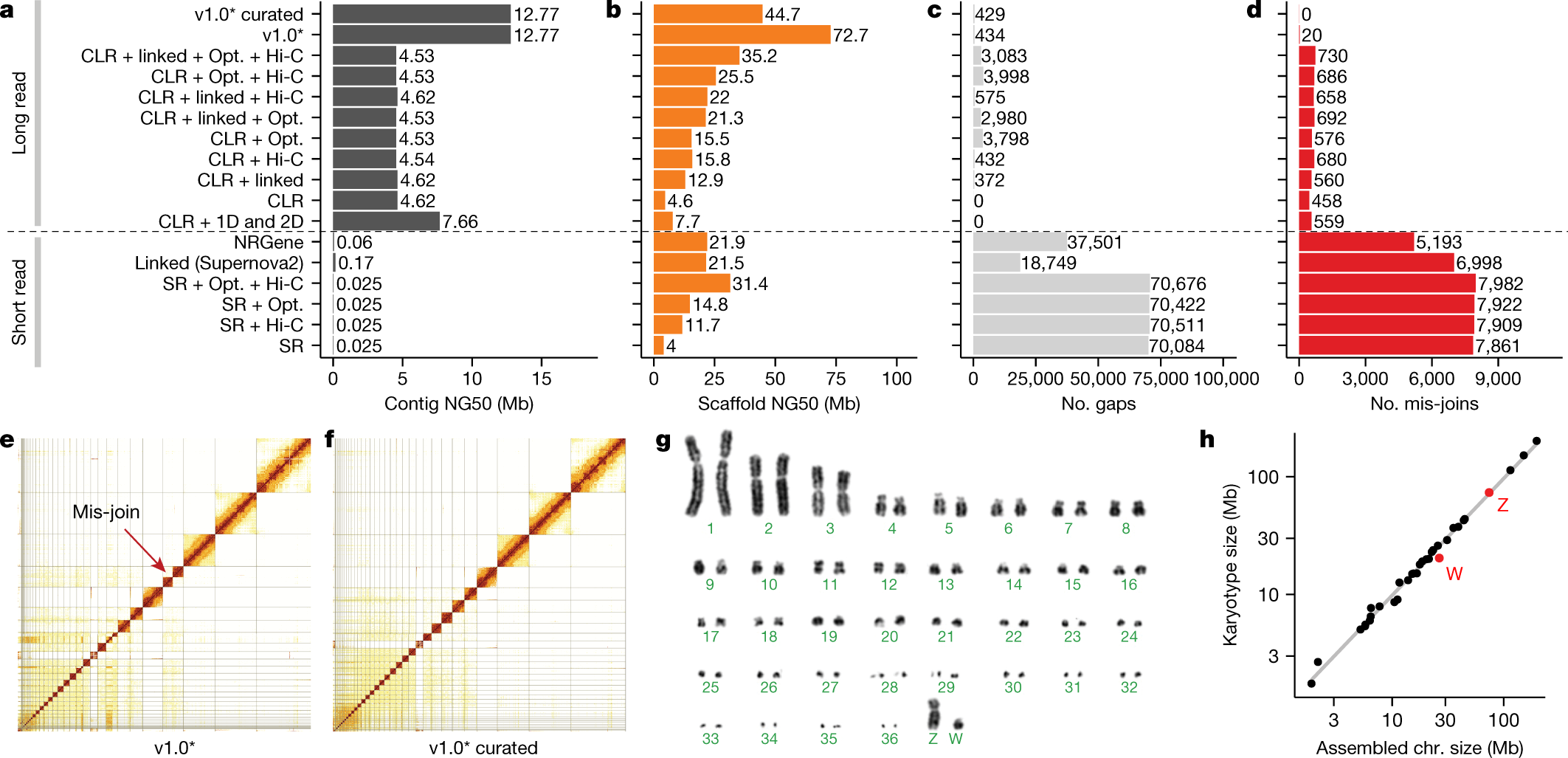 |
|---|
| Fig. 1 from Rhie, Arang, Shane A. McCarthy, Olivier Fedrigo, Joana Damas, Giulio Formenti, Sergey Koren, Marcela Uliano-Silva et al. “Towards complete and error-free genome assemblies of all vertebrate species.” Nature 592, no. 7856 (2021): 737-746. |
Robust haplotype-resolved assembly of diploid individuals without parental data. [arXiv:2109.04785]
Minimizer-space de Bruijn graphs: Whole-genome assembly of long reads in minutes on a personal computer. [DOI:10.1016/j.cels.2021.08.009, bioRxiv:10.1101/2021.06.09.447586]
 |
|---|
| Graphical abstract from Ekim, Barış, Bonnie Berger, and Rayan Chikhi. “Minimizer-space de Bruijn graphs: Whole-genome assembly of long reads in minutes on a personal computer.” Cell systems 12, no. 10 (2021): 958-968. |
Three picks in assembly: one wide (VGP), one deep (hifiasm), one fast (mdBG).
As can be expected of a wide study effort a lot of lessons have been learned and some best practices extracted. Work done by VGP shows that long read sequencing is quintessential for high quality assemblies, but for high quality assemblies a combination of different sequencing approaches is still required. Additionally authors proposed a joint set of 14 metrics grouped into 6 categories as a summary way of characterizing assembly quality. It’s important to note that development of metrics and standards for summary statistics is necessary for development of the field, but caveats should always be explored for any proposed scheme.
For the deep option we picked a recent update to hifiasm that allows haplotype-resolved assembly without parental data. By taking advantage of combined HiFi and Hi-C sequencing data updated version of the tool annotates assembly graph with links inferred from Hi-C data and subsequently uses these links to phase the haplotypes. This work is appealing since haplotype-resolved assembly without the need for parental data can improve the cost of high quality assemblies without sacrificing the detail.
Finally for the fast (and compact too) entry we have mdBG. I have to compliment the use of Rust, because it confirms my suspicion that we are moving past the days when fast and lightweight meant C (although C will never die). Besides blazingly fast and compact (assembly of a human genome at ~50x coverage from HiFi reads in about 10 minutes wall clock time utilizing 10GB RAM running with 8 threads on a Xeon 2.60 GHz CPU) mdBG is also a great example of synthesis of the bioinformatics ideas. Both minimizers and de Bruijn graphs had their place in bioinformatics for a long time, so it is pleasant to see a meaningful merge of the two concepts that brings notable algorithmic advantages. And as a teaser of a very recently posted related preprint on compacted dBGs, Cuttlefish 2 looks like a must read and we cannot wait to dig into the details.
Meta -omics & Microbiome [3 papers]
- Microbiome-based disease prediction with multimodal variational information bottlenecks. [bioRxiv:10.1101/2021.06.08.447505]
 |
|---|
| Fig. 1 from Grazioli, Filippo, Raman Siarheyeu, Giampaolo Pileggi, and Andrea Meiser. “Microbiome-based disease prediction with multimodal variational information bottlenecks.” bioRxiv (2021). |
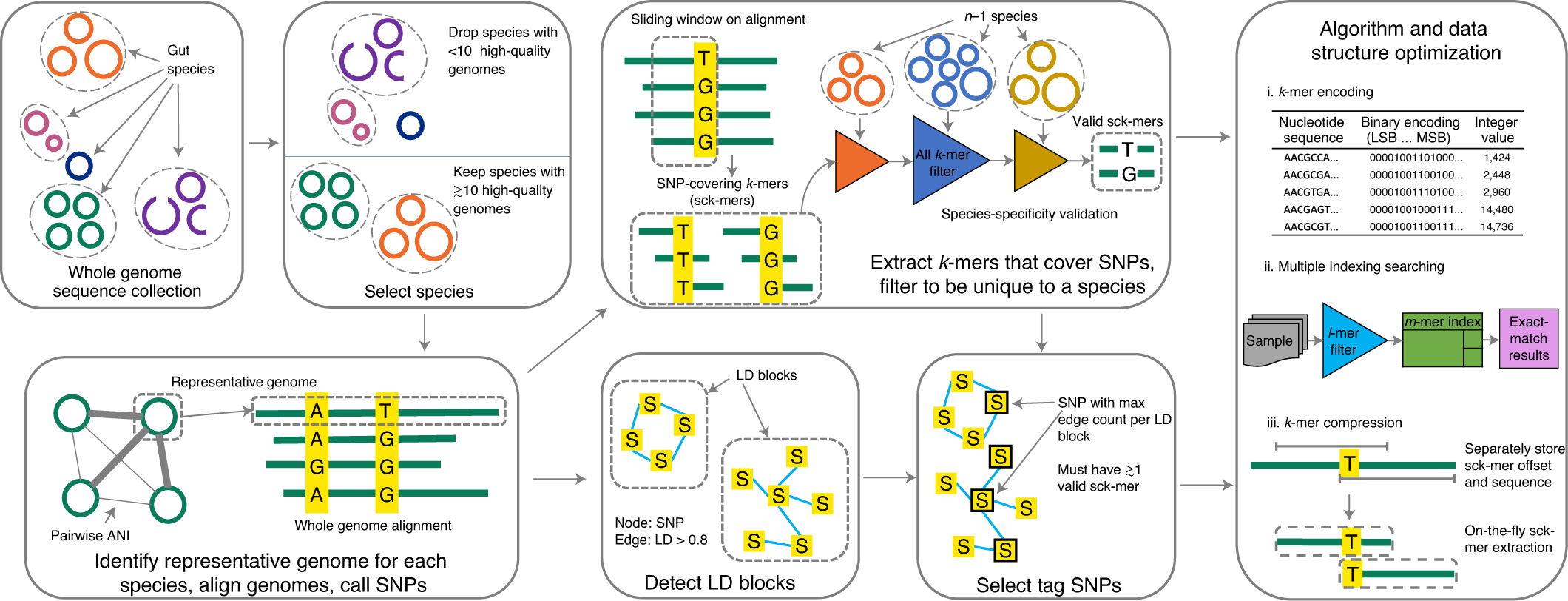
- Fast and accurate metagenotyping of the human gut microbiome with GT-Pro. [DOI:10.1038/s41587-021-01102-3, bioRxiv:10.1101/2020.06.12.149336]
 |
|---|
| Fig. 1 from Shi, Zhou Jason, Boris Dimitrov, Chunyu Zhao, Stephen Nayfach, and Katherine S. Pollard. “Fast and accurate metagenotyping of the human gut microbiome with GT-Pro.” Nature Biotechnology (2021): 1-10. |
- Gramtools enables multiscale variation analysis with genome graphs. [DOI:10.1186/s13059-021-02474-0]
 |
|---|
| Fig. 1 from Letcher, B., Hunt, M. & Iqbal, Z. Gramtools enables multiscale variation analysis with genome graphs. Genome Biol 22, 259 (2021). |
MVIB paper presents a deep learning approach to -omics data integration and subsequent phenotype prediction. Kristen Curry, PhD student in the lab, recently published a review covering this method. Some of the most appealing points in the paper are interpretability of the results, potential for metabolomic data integration, and neat use of transfer learning which is inspired by the biological hypothesis. Overall this work suggests that usage of deep learning in the (meta)genotype to phenotype can be interpretable and efficient while pushing the state of the art performance.
On the flip side, we cannot really talk about the (meta)genotype to phenotype mapping problem if we are stuck getting the metagenotype profile in the first place. GT-Pro relies on k-mers instead of the read mapping for the genotyping of metagenomic samples. Corresponding database of more than 100 million core genome SNPs from 909 bacterial species that are common to human gut presents a valiant effort in systematizing known microbial variation. A reasonable follow up question is whether some form of such databases will become an accepted standard that will be growing and extending beyond human gut related bacteria.
Finally, we arrive back to the topic of variation analysis with genome graphs via Gramtools. Genotyping bacterial genomes typically relies on sequencing short or long reads and mapping them back to a reference genome, or assembly of a bacterial genome combined with whole genome alignment. While the former suffers from reference bias, the latter can be affected by genome assembly quality and genome alignment errors. To address these concerns Letcher et al capture variation by using directed acyclic graphs DAGs that are successions of locally hierarchical subgraphs. It was exciting to see the improved genotyping accuracy of gramtools compared to the popular vg and GraphTyper2 toolkits (improvements specific to large deletions and overlapping small variants in M. tuberculosis). With respect to future work, the gramtools authors state:
“More work is required in the pangenomics field to understand the different real-world performance challenges of repetitiveness (P. falciparum is much more repetitive than human), genome size (microbes are tiny but diverse) and amount, type and density of variation.”
Nick’s pick [1 bonus paper]
I will allow myself a bit of leeway by throwing in a paper that I found really interesting. This is not exactly a lab pick, but something I personally wanted to work into this post.
- The Labeled Direct Product Optimally Solves String Problems on Graphs. [arXiv:2109.05290]
Graph representations of genomes are ubiquitous at this point, but complete theoretical characterizations of the algorithmic complexity bounds on some of the common problems remain un-characterized. In this paper authors introduce an elegant way of characterizing the complexity of common string problems on graphs via a labeled version of tensor graph product construction. The main advantage of the construction presented in the paper is the ability to describe string problems on various graphs while using the same fundamental approach.
Furthermore, since biological graphs exhibit certain local and global properties, it can be fruitful to further analyze questions of possible space compression (e.g. via information theory based proofs) of the graphs that can still efficiently answer relevant string queries. Combined with the prior work on the De Bruijn graph space bounds, this line of research might expand and solidify theoretical characterization of several key problems in computational genomics.
Advait’s fav [Another bonus for it’s New Year’s Eve] {#bonus2.0}.
- MONI: A Pangenomics Index for Finding MEMs. [bioRxiv:2021.07.06.451246v1]
Finding Maximal Unique Matches (MEMs) among a set of genomes has proved to be one of the most effective strategies of read alignments as well as pairwise and multiple alignments. A lot of well-known techniques have been developed to enable efficient identification of MEMs including the FM-index and the Burrows-Wheeler Transform. This paper presents a significant advancement in a slew of recent papers that improve index construction an MEM retrieval.
MONI builds on the previous work that showed the best way to sample O(r) entries of the SA such that locating queries is efficient using prefix-free parsing (PFP) called r-index. The main contribution of the paper is an efficient way to compute matching statistics (through a data structure called thresholds) that enables finding MEMs using a r-index. MONI when compared to popular read aligners used 2–11 times less memory and was 2–32 times faster for index construction which is a notable advancement in the field.
Dr. Nicolae Sapoval
PhD student
Dr. Sapoval received his Ph.D. in Computer Science from Rice University in May 2024. Previously he obtained a B.S. degree in Computer Science and a B.S. with Honors in Mathematics from the University of Chicago. At the University of Chicago Nick worked in wireless networks research and later in computational biophysics focusing on conformational transition modeling for insulin degrading enzyme. His current interests are in the areas of computational biology with a focus on genomic data.
Dr. Advait Balaji
PhD student from 2018 through 2023 (currently Analytics Engineer at Oxy)
Advait (5th year PhD student) obtained a dual degree, B.E Computer Science and MS Biological Sciences from BITS, Pilani in India. During his undergraduate degree, he received the Khorana Scholarship (2016) from the Indo-US Science and Technology Forum and also a thesis fellowship (2017-18) to work at Icahn School of Medicine, Mount Sinai, NY. At Mount Sinai, he worked on creating a Sub-cellular process-based ontology that predicts whole cell function using Natural Language Processing. His research interests are at the intersection of genomic data science and designing efficient algorithms to analyze genomic data.