Elucidating the Rules of Cooperation and Resiliency in Microbial Communities through Stochastic Graph Grammars
Overview
Microbial communities in aquatic ecosystems are central to maintaining the resiliency of these important environments. These dense microbial populations are rich in species diversity and their genomes are dynamic because of processes such as gene duplication and horizontal gene transfer. Despite extensive genomic flux due to these evolutionary processes, microbial communities cooperate and maintain stable interactions and are resilient to environmental perturbations. This project aims to develop a suite of computational tools for deciphering the rules that govern cooperation and resiliency in hot spring microbial mats, or biofilms, from Yellowstone Park. These well-studied environments are ideal for examining how critical environmental parameters such as temperature, light and nutrients influence the diversity, abundance, and evolution of microbial populations. Novel computational tools will be developed to identify rules that govern relevant biological processes. Understanding how microbiomes evolve and adapt, especially with respect to the environment and to perturbations is crucial to understand many aspects of life. Thus, the results obtained from this project will have a significant impact on biology, health, conservation efforts, and animal and plant management. Furthermore, this research will support the interdisciplinary development of a diverse cohort of PhD and undergraduate students at Rice University and Carnegie Institution for Science located on the Stanford University campus. This project will also support the development of a summer REU focused on bioinformatics, ecology, evolution, and microbiology, and will also include summer training opportunities for graduate students with collaborators at the Pasteur Institute in Paris, France. This collaborative, multidisciplinary project presents an innovative plan for combining education and outreach activities to achieve real and lasting impact. All software developed will be made available on open-source code repositories.
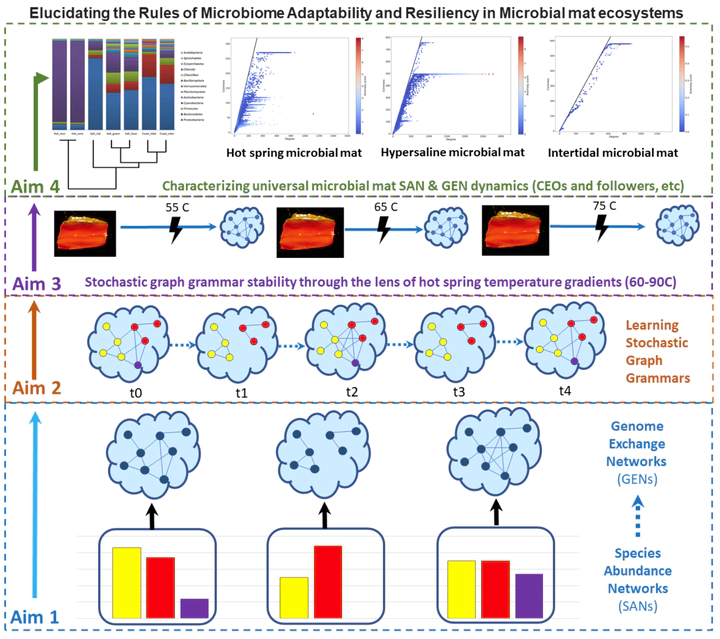
The overall goal of the project is to understand how microbiomes evolve and adapt, especially with respect to the environment and to perturbations. To meet these goals, the team of researchers will introduce methodological advances at the intersection of graph theory, formal languages, and machine learning to identify rules governing gene duplication and horizontal gene transfer from metagenomic sequencing data. In Aim 1, the project will construct and correlate species abundance networks with genome exchange networks through genome assembly graphs. In Aim 2, the project will produce a stochastic graph grammar framework for modeling network evolution over time, as well as unveil new rules with deep graph generators. In Aim 3, genome-level simulations will be combined with the genomic events identified in Aims 1 and 2 to assess the evolutionary benefits of these biological processes and their associated rates. The final project aim will focus on characterizing cooperation and resiliency through the study of graph grammars and network analysis techniques. The open-source computational methods developed across all four project aims will be evaluated on four distinct metagenomic datasets, including both environmental and host-associated microbiomes. Finally, a wide variety of metagenomic datasets will be used to show generalizability of the computational approaches to host-associated microbiomes.
Co-PIs
- Dr. Devaki Bhaya (Carnegie Institute)
- Dr. Santiago Segarra (Rice University)
- Dr. Luay Nakhleh (Rice University)
Collaborators
- Dr. Eduardo Rocha (Institut Pasteur)
Dr. Kristen Curry
PhD student
Dr. Curry received her Ph.D. in Computer Science from Rice University May 2024. She obtained a bachelor’s in computer science from the University of California, Berkeley. Prior to enrolling at Rice, she worked for a private biotech company focused on generating personalized health information from blood protein levels. Her primary research interests are microbial interactions and their impact on host health. Outside of the lab, she enjoys backpacking, running and practicing yoga.
Dr. Michael Nute
Research Scientist
Mike (Research Scientist) received his Ph.D. in Statistics in 2019 from the University of Illinois at Urbana-Champaign where he was advised by Dr. Tandy Warnow in the Department of Computer Science and worked on algorithms related to multiple sequence alignment and phylogenetic tree estimation, in particular applying these methods to studying microbial communities. He was co-advised by Dr. Rebecca Stumpf in the Department of Anthropology where he and other lab members developed novel methods to compare the microbiomes of human and non-human primates. His research interest is in discovering a new applications for our understanding of microbial communities.
Todd J. Treangen
Associate Professor of Computer Science, Bioengineering
My research interests include algorithms and data structures for efficient analysis of microbial genomes and metagenomes