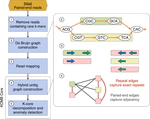
Characterizing metagenomic samples via kmer-based, database-dependent taxonomic classification methods has provided crucial insight into underlying host-associated microbiome dynamics. However, novel approaches are needed that are able to track microbial community dynamics within metagenomes to elucidate genome flux in response to perturbations and disease states. Here we describe KOMB, a novel approach for tracking homologous regions within microbiomes. KOMB utilizes K-core graph decomposition on metagenome assembly graphs to identify repetitive and homologous regions to varying degrees of resolution. K-core performs a hierarchical decomposition which partitions the graph into shells containing nodes having degree at least K, called K-shells, yielding O(V + E) complexity compared to exact betweenness centrality complexity of O(V E) found in prior related approaches. We show through rigorous validation on simulated, synthetic, and real metagenomic datasets that KOMB accurately recovers and profiles repetitive and homologous genomic regions across organisms in the sample. KOMB can also identify functionally-rich regions in Human Microbiome Project (HMP) datasets, and can be used to analyze longitudinal data and identify pivotal taxa in fecal microbiota transplantation (FMT) samples. In summary, KOMB represents a novel approach to microbiome characterization that can efficiently identify sequences of interest in metagenomes.